Designing Proteins for Dummies
5 min readLast week, I decided to participate in Adaptyv’s Protein Design competition. Having some vague exposure to protein models on Twitter, I thought it would be helpful to actually try my hand at designing a protein from scratch. The competition was a good excuse / incentive to invest the time into learning how it worked under the hood.
How do people use protein models?
In 2023, we saw the first generative protein models, which were able to generate new proteins from scratch, just like how an LLM generates text. These models are generally diffusion-based, like many of the image generation models such as Stable Diffusion. The most famous of these models is RFDiffusion, which was created by Nobel Laureate David Baker and his lab. RFDiffusion takes a protein structure as an input, and it generates another protein that attaches to the input, called a binder. RFDiffusion doesn’t immediately generate a fully formed 3D structure though – it only generates a backbone, which is the “basic structure” of a protein.
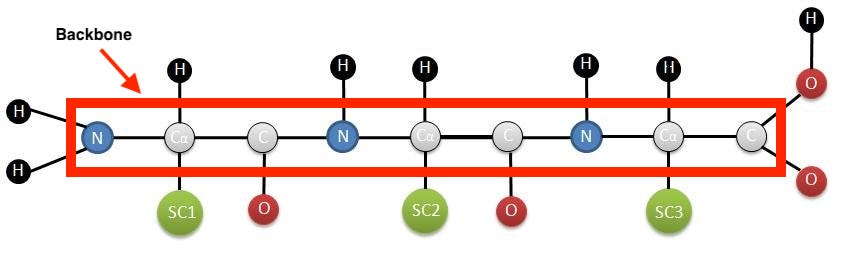
To get the full structure of the protein, we use a different protein model called ProteinMPNN, which “fills in” the backbone with compatible amino acids that adhere to the backbone that was provided as input. ProteinMPNN lets us go from 3D backbone → string of amino acids, like “SIDERWWTTD…”, which is also known as a protein sequence.
Finally, we use AlphaFold (AF) to check the validity of this sequence. AF takes a protein sequence and outputs a 3D structure of that protein, as well as a confidence score. People use AF to check how “valid” a protein sequence is, because if it outputs a 3D structure with very low confidence, we have a strong reason to believe that the original sequence was implausible in the first place.
Hence, the workflow generally goes like this:
- RFDiffusion to generate protein backbone
- ProteinMPNN to generate sequence from the backbone
- AlphaFold to get the structure of the protein, as well as its “validity”
Since these models could hallucinate all kinds of results, scientists usually run this workflow hundreds or thousands of times until they get a good score from AF, which implies a plausible protein structure in reality. Then they will test it in a wet lab to see if it actually works in real life.
My Experience
The workflow that I mentioned was somewhat annoying to set up because I needed to run 3 different models, and write scripts to take the outputs → convert to specific formats before passing it back as inputs into the next model, and so on.
I stumbled upon some new tech called BindCraft that was released 3 months ago, which massively simplified the entire workflow. BindCraft is a pipeline that chains together multiple models and automatically executes the generate → validate loop with as many iterations as the user specifies. Given I was short of time to get a submission in for the competition, I decided to just use BindCraft to generate results.
The goal of the competition was to design a binder capable of neutralizing the Nipah virus, a pathogen with up to 75% mortality rate and high pandemic potential.
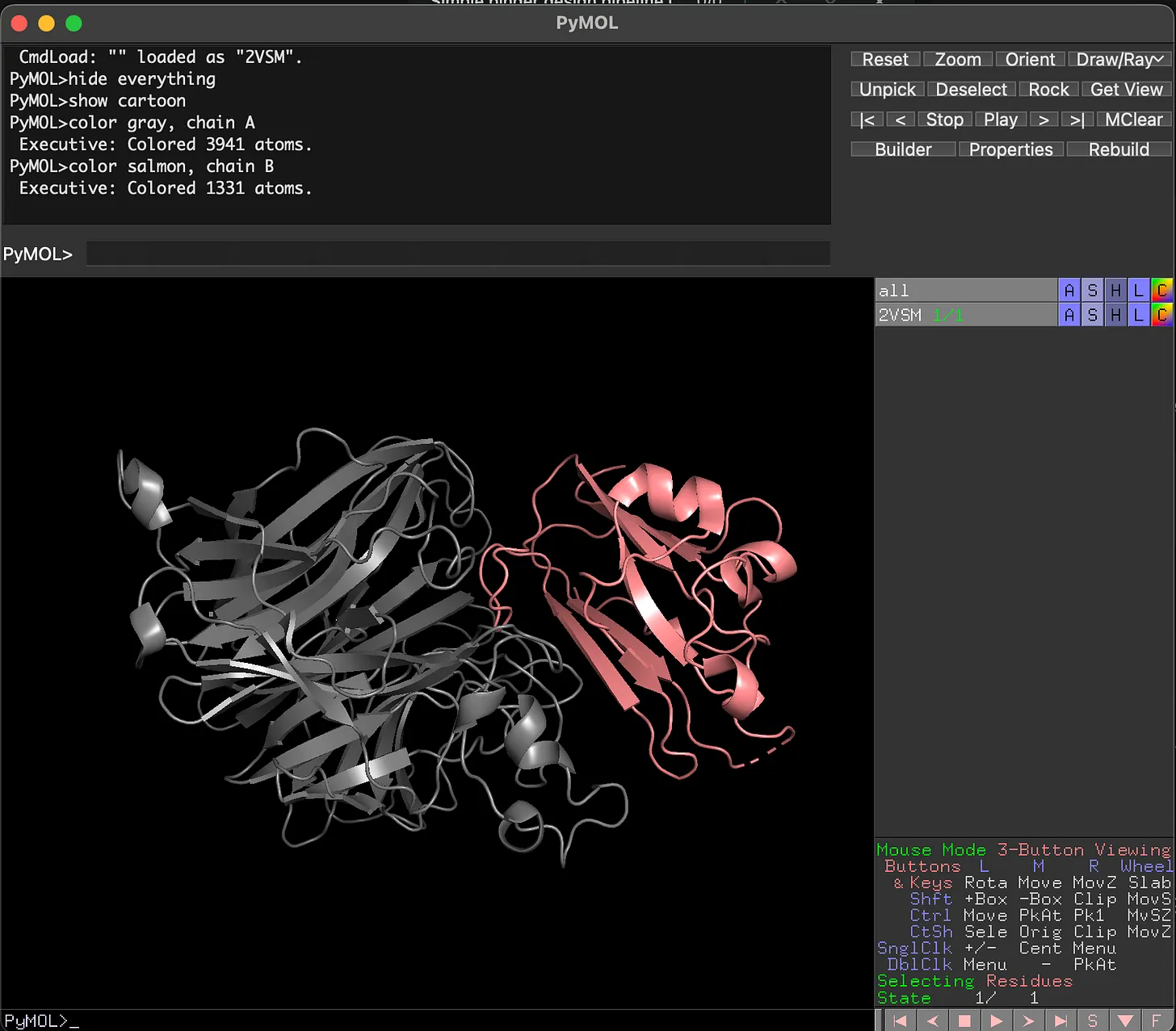
First, we load up the target in PyMOL. As we can see, there are 2 distinct structures here – the gray structure is the virus, whereas the pink structure is the human receptor that the virus binds to within humans. We want to design a new version of the pink structure that can bind to the virus, so that it doesn’t interact with the human cells.
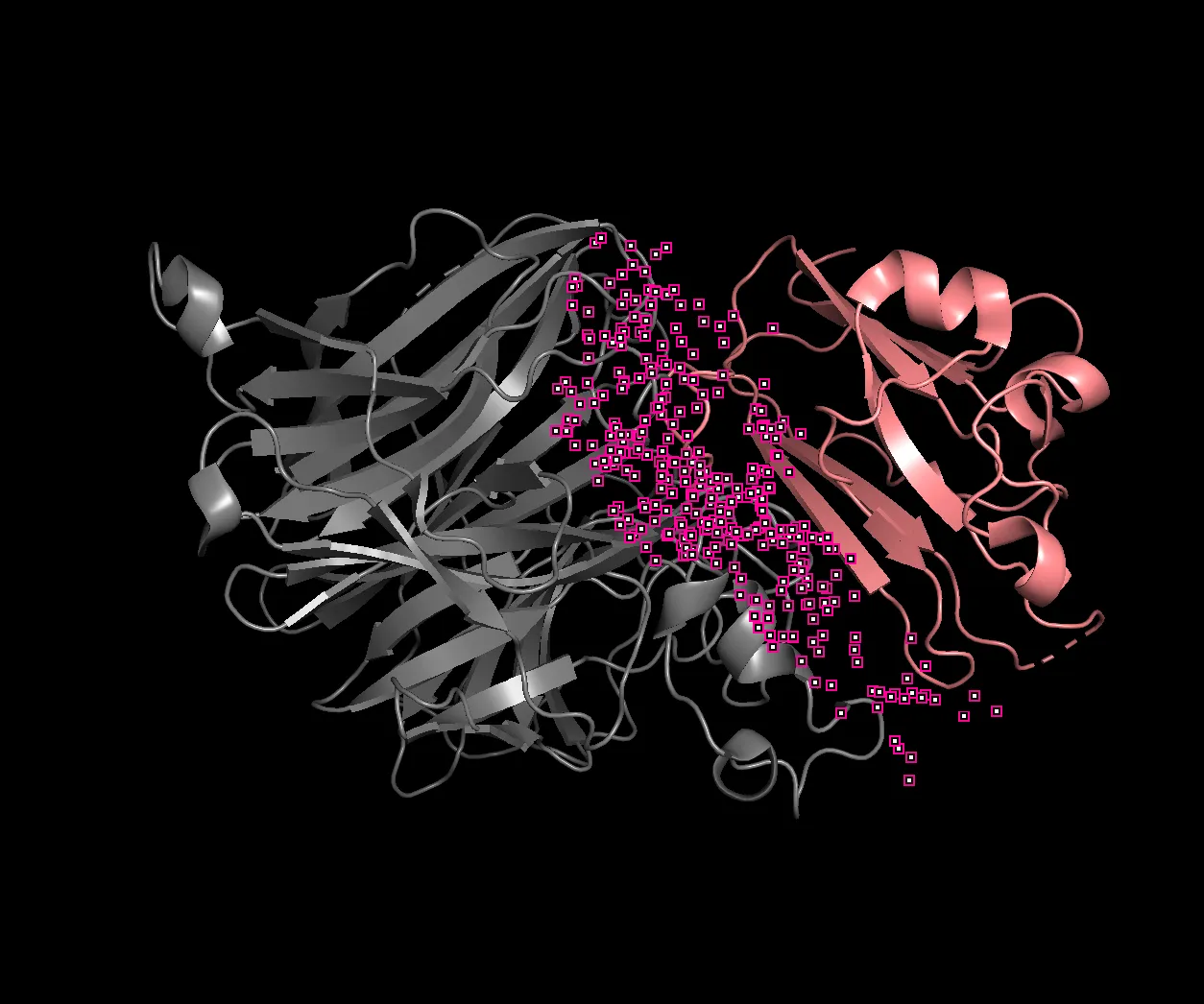
Within PyMOL, we can find the hotspots where the two proteins “touch”, and their locations within the sequence. After running some commands in PyMOL, we get the indices of where the hotspots are within the first (gray) protein.
{
"design_path": "/workspace/BindCraft/NiVG/",
"binder_name": "NiVG",
"starting_pdb": "/workspace/BindCraft/targets/NiVG.pdb",
"chains": "A",
"target_hotspot_residues": "A214,A215,A216,A238,A239,A240,A241,A242,A302,A304,A305,A388,A389,A401,A402,A403,A458,A488,A489,A490,A491,A492,A501,A502,A504,A505,A506,A507,A528,A529,A530,A531,A532,A533,A554,A555,A556,A557,A558,A559,A579,A580,A581,A583,A585,A588",
"lengths": [80, 120],
"number_of_final_designs": 100
}
Next, I define the parameters in which I want the model to run, such as the hotspots, the target protein structure, the length of the binder I want to generate, and the number of iterations… and then I wait.
Results
On an RTX 5090, each “iteration” took approximately 20-30 minutes to run. The results get conveniently saved into a csv file that looks like this.
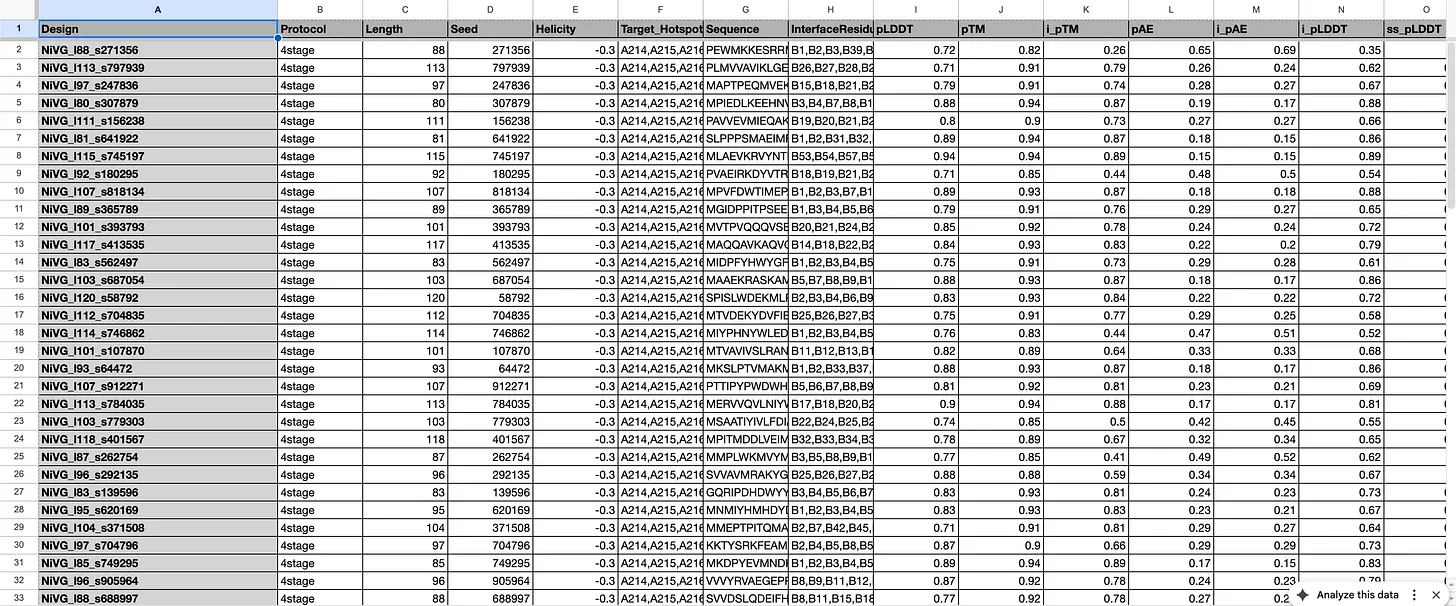
The columns pLDDT, pTM, i_pTM, etc. are all various forms of confidence scores – such as confidence on the structure, confidence of the “fit” between the binder and the target, and so on.
Instead of just submitting the top scorers from this list, I evaluated my ~150 designs using another model to come up with an alternate set of scores. Then, I wrote a script to create an aggregate score which combined both sets of scores (Boltz2 and AF2) and selecting the top scorers on both, in the hopes of selecting more robust designs.
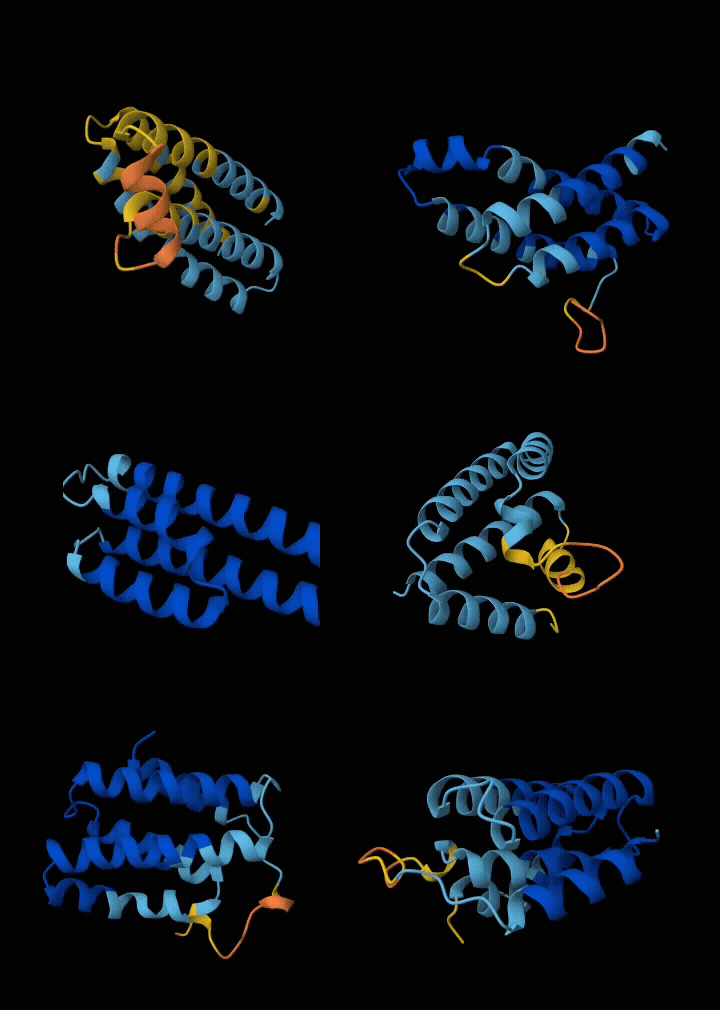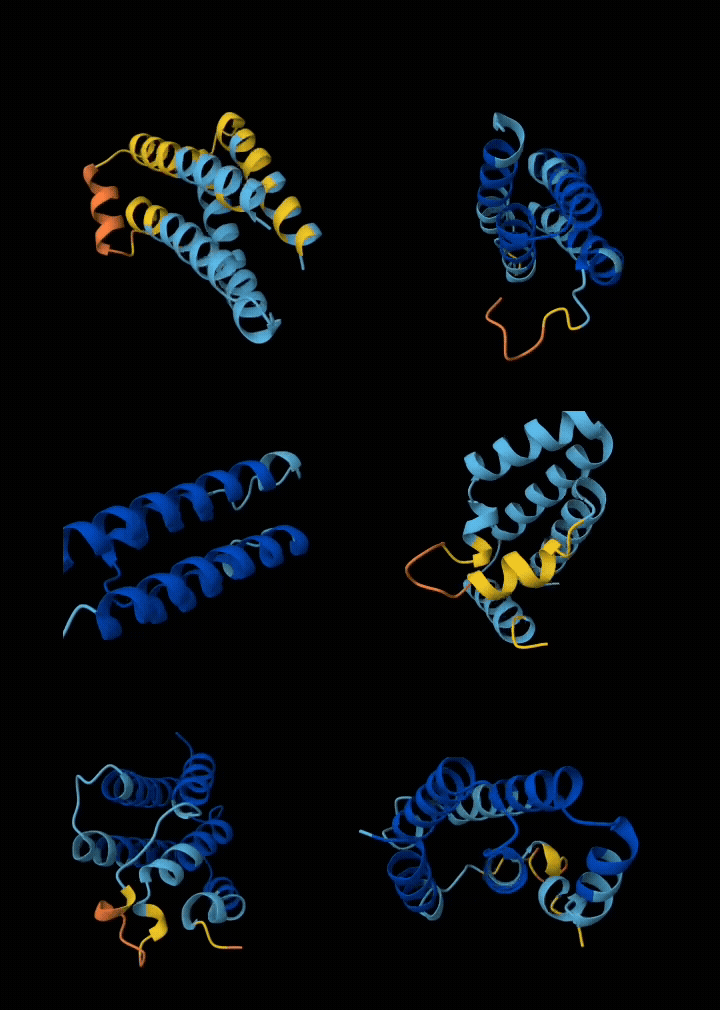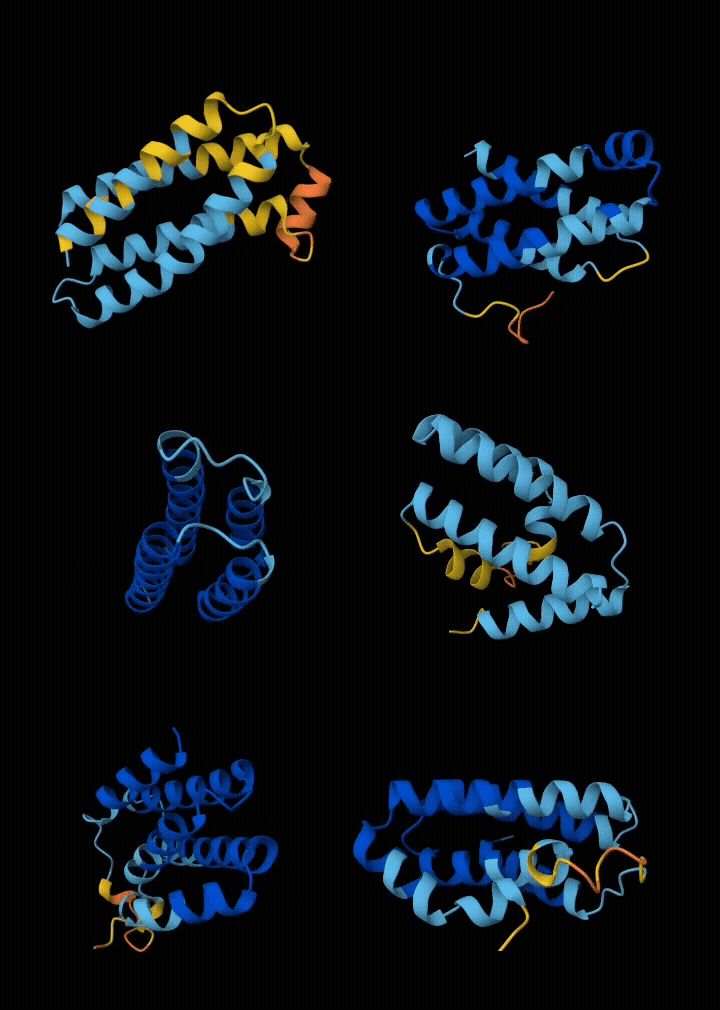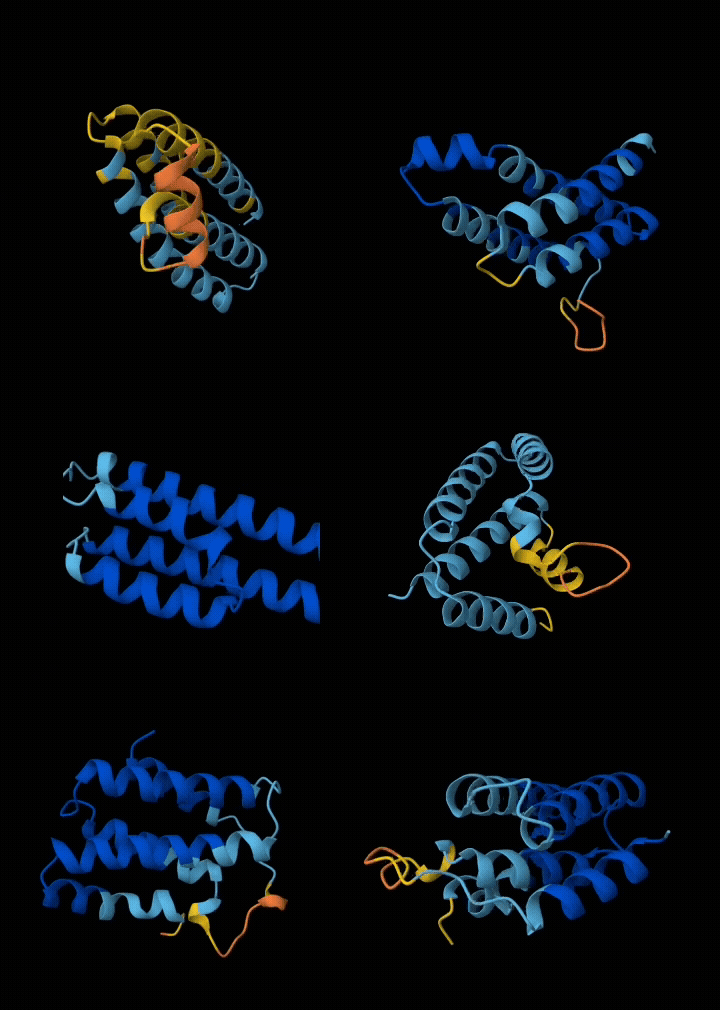
Eventually I narrowed it down to ~6 submissions. If my submissions get selected, they will get tested in a real wet lab to see if they are structurally sound, and if they bind with the target. All in all, I spent ~$200 in compute and a week of my time to get these 6 squiggly things.